技術情報
新しく導入した機器や実験技術のTipsなどを掲載いたします。実験研究にお役立ていただけると幸いです。
リアルタイムPCR | 実験技術のTips|
リアルタイムPCRによる 遺伝子発現の定量について
- PCR増幅曲線の立ち上がりサイクル数は試料中の鋳型量を反映している
- PCR効率の求め方
- リアルタイムPCRで遺伝子発現量を定量するために
- 実際の実験手順
- Oligo(dT)とランダムプライマー
- プローブ法とサイバーグリーン法
PCR増幅曲線の立ち上がりサイクル数は試料中の鋳型量を反映している
PCRは1サイクル毎に鋳型DNAのコピーを合成する反応です。1サイクルで2倍、2サイクルで4倍、3サイクルで8倍に増幅されます。理論とおりであれば20サイクルで1,048,576倍に40サイクルでは1,099,511,628,000倍に増幅されます。 したがってある一定量のDNA量に達するまでのサイクル数が分かれば初期鋳型量が分かります。
しかしながら理論どおりに増幅されない場合もあります。代表的な原因を示します。
- プライマーが100%アニールしない。
- DNAポリメレースに影響を及ぼす共雑物の存在。
1.はPCR条件変更で対応できます。それでも改善されない場合は配列を見直すことになります。2.はサンプルの精製を行うか希釈して共雑物の濃度を下げます。
上記の原因が解決されてもPCR反応が進んで生成物量が増加するにつれて増幅効率は減少し、最終的にはプラトーに達します。その要因として代表的なものは
- ポリメレースの失活
- dNTPの枯渇
- プライマーの枯渇
- 反応副産物であるピロリン酸による合成反応阻害
- 生成したDNA鎖同士の再会合によるプライミング阻害
などが考えられます。これらは全てPCRの後半になるにつれ顕著になります。
リアルタイムPCRは理論どおりにPCR反応が進んでいることが前提ですので増幅効率(PCR効率)を確認することが必須となります。
PCR効率の求め方
通常qPCRは定量範囲を確認するために、事前に既知または高発現のcDNAを用いて幅広く希釈系列を作り、定量範囲を確認する必要があります。また試料を用いたqPCRの結果が定量範囲内にあることが必須条件です。それでも定量範囲確認実験と試料のqPCRが同じ精度で再現性があるという仮定に基づいています。 我々はこの仮定が成り立つかどうかについて不安を持っています。たとえ抽出方法が同じであったとしても共雑物やその濃度が同じであるとは限りません。その共雑物がリアルタイムPCRに影響を与えないという保障もありません。実際cDNA濃度が高い時、立ち上がりサイクル数が遅くなり、希釈することで解消する場合があります。このような不安から我々は全てのPCR効率を求め、確認後、定量計算することを提唱しています。
PCRは初期鋳型DNA量を[DNA]0、増幅効率をe、サイクル数をcとすると
[DNA]=[DNA]0(1+e)c
で表されます。e=1の時( )の中は2になりDNA量が2倍に増えることになります。
上記式の対数を取ると
log[DNA]=log[DNA]0+log(1+e)c
=log[DNA]0+clog(1+e)
PCR産物が一定量に達するサイクル数をCtとすると
Ct=(log[DNA]t-log[DNA]0)/log(1+e)
となります。Ctはlog[DNA]0の関数で表され傾きは-1/log(1+e)となります。
傾きの理論値は-3.3219になります。
PCR効率(増幅効率)は下記の式で求められます。
増幅効率 e=10(-1/傾き)-1
(EXCELでは"=POWER(10,-1/E10)-1"と入力します)
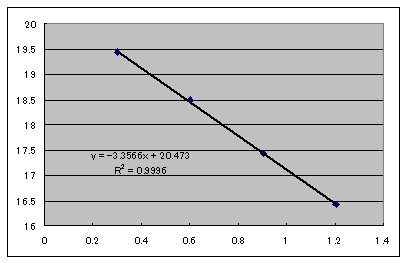
実際の実験ではサンプルの希釈系列を作りリアルタイムPCR測定を行って立ち上がりのサイクル数を求めます。横軸に鋳型DNA濃度の対数をとり、縦軸に立ち上がりのサイクル数をとると直線関係が得られます。鋳型となるDNA濃度は不明なことが多いため濃度比を使います。
一例を示します。下図はマウス組織から調整したcDNAを20倍,40倍,80倍,160倍希釈し、GAPDHをTaqMan法で測定したものです。横軸には便宜上20倍希釈を16、40倍希釈を8、80倍希釈を4、160倍希釈を2としてその対数を取りました。傾きが-3.3566ですのでPCR効率は0.9857となります。
検量線(PCR効率グラフ)の解釈
検量線の縦軸は増幅曲線の立ち上がりサイクル数、横軸は目的遺伝子(cDNA)の濃度比を常用対数としたものです。理想は、得られた各遺伝子の近似式、全てでR2乗値が1.0、PCR効率が理論どおりであることです。この場合、全ての検量線は平行関係となり、違うのは縦軸(切片)となります。この縦軸の差(ΔCt)が各遺伝子間の存在比を示しています。実際に求めたPCR効率は理論値±5%を良しとしています。5%の差であっても30サイクルでは2倍以上の差となることから、PCR効率は理論値±5%でΔCtが1以上の差が認められた場合のみ有意と判断します。
リアルタイムPCRで遺伝子発現量を定量するために
- 仮定1 一定量の細胞から遺伝子抽出を行うこと
- 仮定2 遺伝子抽出の効率が常に一定であること
- 仮定3 cDNA合成反応が常に一定であること
- 仮定4 リアルタイムPCR効率が常に一定であること
以上の仮定が全て成り立てばリアルタイムPCRによって遺伝子発現量を定量することが出来ます。
仮定1について 実際の試料で厳密に細胞数を合わせることは不可能です。 仮定2について 効率が常に一定であることはありえません。
そこで仮定1、2を補正するために内在性コントロール遺伝子(β-アクチン、GAPDH、18SrRNAなどでHouse keeping geneとかEndogenous geneといいます)を同時に測定し、内在性コン トロール遺伝子と目的遺伝子の発現量の差を求め、サンプル間の目的遺伝子発現量を 相対的に比較します。この場合、内在性コントロール遺伝子は細胞当り等しく発現しており、細胞の状態には依存しないことと、内在性コントロール遺伝子と目的遺伝子の抽出・測定効率に差が無いことが前提となります。。
私たちは2ステップ法を推奨しています。まず新鮮な試料からTotal RNAを抽出します。もし試料のまま保存する場合は、市販の保存液(RNAlatorなど)に浸けて低温で保存してください。抽出操作はRNAの分解を最小限にするため、使う試薬や器具、消耗品などは全て専用のものを使うこと、試料量が少ない場合は必ずキャリアーとしてグリコーゲンを添加すること、比較的安定なステップ(アルコール沈殿)まではできるだけ速やかに実験をおこなう事が大切です。また、できるだけ早くcDNAを合成し分注して冷凍保存します。
常にPCRの増幅効率を求め確認すれば、最初に示した仮定4を満たしたことになります。また内在性遺伝子との差から遺伝子発現量を求めることで最初の試料の差(仮定1)や抽出効率の差(仮定2)を補正することができます。さらに同時に測定することにより希釈などのピペッティングの誤差を最小限にすることができます。仮定3のcDNA合成効率については目的遺伝子と内在性遺伝子間で差が無ければ(差を無視できれば)良いのですが私たちには解決策が見つからなかったので、キットと自分の実験技術を信じて実験をおこなっています。詳細についてはお問い合わせください。
検量線からCt値を求める我々の流れは、まず得られた近似式が評価に耐えられるかどうかをR2乗値と傾きから求めたPCR効率で評価し、評価できると判断した場合に、その切片(Ct値)を用いて定量計算を行います。一見手間が掛かりそうですが、希釈系列を作るか1点希釈とするかの違いです。また費用も1サンプル1遺伝子あたり4ウェルなので大きく変わりません。4ウェルは最低必要で余裕がある場合は6ウェルとします。
解析に少々時間が掛かりますが、各サンプル、各遺伝子、全てでPCR効率を確認できるメリットは大きいと思います。何より得られたデータの信頼性に自信を持てることが最大のメリットです。
The MIQE Guideline : Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clinical Chemistry 55:4,611-622(2009)よりTable 1のMIQE checklist for authors, reviewers, and editorsから
qPCR validationの必須項目を書き出してみました。(この論文ではCtをCqと表現します)
1)Specificity (gel, sequence, melt, or digest)
2)For SYBR Green I, Cq of the NTC (no-template controls)
3)Calibration curves with slope and y intercept
4)PCR efficiency calculated from slope
5)r2 of calibration curve
6)Linear dynamic range
7)Cq variation at LOD ( limit of detection)
8)Evidence for LOD
9)If multiplex, efficiency and LOD of each assay
我々の手順により3)から9)までの項目はクリアできます。
実際の実験手順
私たちが行っているリアルタイムPCR実験では、cDNA溶液をミリQ水で20倍に希釈し、更に倍(40倍希釈)、更に倍(80倍希釈)、更に倍(160倍希釈)と最低4点の希釈系列を作ります。発現が多い遺伝子の場合はもっと希釈したところから出発しても良いでしょうし、逆に発現が少ない遺伝子の場合は10倍希釈から始める事もあります。貴重なcDNAの場合や測定する遺伝子の数が多い場合は、一回の実験に使うことが出来るcDNA量が限られますので、希釈倍数で調節することになります。その溶液を測定する遺伝子の分(内在性コントロール遺伝子と目的遺伝子を足した数)だけ分注します。私たちは分注誤差を考えて4ulずつ分注しています(定量精度を上げるためのコツ)
。例えば、4点希釈で、目的遺伝子と内在性遺伝子で8種類の遺伝子を分析する場合、それぞれの遺伝子につき160倍、80倍、40倍、20倍希釈のcDNA溶液が必要ですので合計32本となります。96ウェルプレートでは3種類の試料について、それぞれ8種類の遺伝子を測定できることになります。 つぎに遺伝子ごとのミクスチャーを調整し、6ulずつ加えて、軽く混合して測定します。
「測定例」をご参照ください。テンプレートファイルはこちらから。
測定終了後に解析を行います。このとき各遺伝子のスレッショルドレベルを常に一定の値として、スレッショルドサイクルCtを求めます。ベースラインの設定は増幅曲線を見ながら最適の値に設定します。実験条件と解析条件を常に同じにしないと、別のときに行った結果との比較ができなくなります。
測定終了後の解析方法について補足します。
スレッショルドレベルの設定について
測定系(PCR条件を含め)が同じであればテンプレートの濃度に係わらず蛍光が検出できない始めの部分と、産物がこれ以上増えずプラトーに達した蛍光強度はほぼ一定になります。PCRはサイクルが増えるにしたがって効率が悪くなるため、出来るだけ低い値に設定します。 この値は遺伝子ごとに変えても良いですが、一連の実験で同一遺伝子のスレショルドレベルは同じにします。これによって複数のプレートに亘ってサンプル間の比較が可能になります。
ベースラインの設定について
データからスレッショルドサイクル(スレッショルドレベルと増幅曲線の交点のサイクル数)を求めるにあたって手動で設定する項目は、上記のスレッショルドレベルとベースラインになります。スレッショルドレベルは一連の実験においては同じに設定しますので、実験者が設定できる項目はベースラインだけになります。解析ソフトで自動設定も出来ますが、増幅曲線とスルッショルドサイクルを確認しながら手動で設定します。自動設定では各ウェル毎にstartと endが設定されますが、手動では希釈系列(4点以上)を確認しながらPCR効率を意識して設定します。その際、近似式を得るためには最低3点が必要です。言い換えれば4点の内1点は外しても近似式は得られるということです。
得られたCt値を使って表計算ソフトでグラフを書き、その近似曲線から遺伝子発現量を算定します。横軸にテンプレート濃度(私たちは便宜的に16、8、4、2としています)の対数(倍倍希釈ですから、等間隔となります)、縦軸にCt値をとると理論的には傾きは-3.3219、R2=1、切片はその遺伝子の量に相関した値となります。私たちは傾きが±0.1(PCR効率にすると±5%)、R二乗値は0.98以上を原則としています。この値から外れた場合は再解析か測定をやり直すことになります。そして内在性遺伝子と目的遺伝子のCt値の差(ΔCt:切片の差 [Target gene Ct]-[Endogenous gene Ct])を求め、試料間のΔCtの差(ΔΔCt)から遺伝子発現の量比を計算します。 (EXCELでは"=POWER(2, -『ΔΔCtの値』)"と入力します)
「リアルタイムPCRの結果解析例」をご参照ください。
Oligo(dT)とランダムプライマー
リアルタイムPCRではOligo(dT)かランダムプライマーを使ったcDNA合成が一般的です。 もし合成したcDNAをリアルタイムPCRしか行わないのであればランダムプライマーをお勧めします。Oligo(dT)の場合、ポリAにアニールしますので全長のcDNAが期待できます。したがってクローニングやノーザンブロットには良いのですが、アニールサイトが限定されるため必ずしも全てのポリA-RNAに対してcDNAが出来るわけではありません。またポリAサイトから遠く離れた部分までcDNAが合成できるとも限りません。ランダムプライマーの場合はポリA-RNAに限らず一定以上の長さがあればアニールサイトが存在しますので断片化されたcDNAが出来ることになります。断片化といってもリアルタイムPCRでの産物よりも十分に長いので支障が出ることはありません。むしろポリAの位置に関係なくcDNAが出来るのでプライマーの位置を気にする必要もなく便利です。経験的にはランダムプライマーの方が数サイクル早く立ち上がる印象があります。
プローブ法とサイバーグリーン法
プローブ法とはフォワードプライマーとリバースプライマーの間にアニールするようにプローブを設計します。プローブは両端に蛍光色素とその蛍光を消光する物質でラベルされており、PCRによってポリメレースが相補鎖DNAを合成しながら進行方向にアニールしたプローブを分解することによって蛍光色素がフリーとなり蛍光検出されます。プライマーとプローブが共にアニールし、しかもプローブが分解されて始めてフリーの蛍光が出ることから特異性が高いことになります。主だった生物種、殆んどの遺伝子に対して購入することが出来ますので、条件検討に時間を避けない場合や多くの遺伝子発現を見たいときなどはプローブ法をお勧めします。
サイバーグリーン法はサイバーグリーンを加えてPCRを行うため、2本鎖DNAが出来れば蛍光を持ちます。したがってプライマーダイマーや目的産物以外のDNAも検出されてしまいます。そのためサイバーグリーン法を行う場合はリアルタイムPCR終了後、徐々に温度を上げていき2本鎖DNAが1本鎖に乖離する温度を確認することで産物の純度確認を行う必要があります。もしこのときに目的以外のDNAの存在が確認された場合はPCR条件の変更や、プライマーを設計し直す必要があります。サイバーグリーン法の利点は高価なプローブを用意しなくても良いことと感度が高いことです。プローブ法の場合は分解したプローブの分だけ蛍光物質が放出されますが、サイバーグリーン法では2本鎖DNAの数と長さに比例してサイバーグリーンが蛍光を持つため、感度が1桁から2桁高くなります。
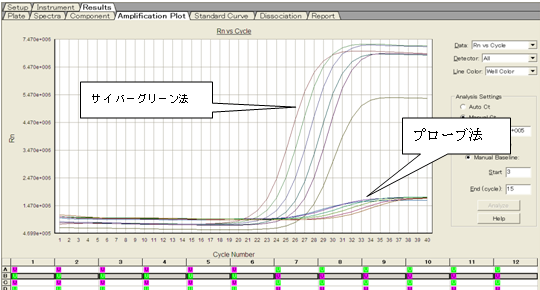
比較実験結果を示します。
Templateはmouse brain cDNA、40倍から倍倍希釈し使用。
Rodent GAPDHのprimer濃度とprobe濃度を変えて実験。
probeなしの場合はSYBR Green法、probe入りの場合はTaqMan法で分析。
Rodent GAPDH (TaqMan) |
||||||
| primer | 100nM | 100nM | 100nM | 200nM | 200nM | 200nM |
| probe | - | 100nM | 200nM | - | 100nM | 200nM |
| PCR効率 | 0.96459183 | 0.97235575 | 0.99846993 | 1.01332746 | 1.01131886 | 1.00097247 |
| 切片 | 21.71 | 28.136 | 26.466 | 19.952 | 25.444 | 24.295 |
Primer、probe共に100nMよりも200nMの方が、立ち上がりが早くなっています。
プローブ法よりもサイバーグリーン法のほうが早く立ち上がっています。